
About
Our lab aims at understanding how genetic changes between individuals can or cannot result in disease by quantifying the impact mutations have on protein aggregation and toxicity.
We are particularly interested in amino acid sequences that can adopt different conformations and undergo a process of self-assembly which results in distinct physical states.
The aggregation of proteins into insoluble amyloid fibrils is a key process in the pathogenesis of a number of neurodegenerative conditions, such as Parkinson’s disease or Amyotrophic Lateral Sclerosis. However, examples of functional amyloid are also widespread in nature, especially across bacteria and fungi. Our work aims at systematically deciphering the sequence-dependencies of the process of aggregation in both functional and pathological contexts.
Recently, it has become clear that proteins can also self-assemble into a more dynamic and reversible state through a process of liquid de-mixing which is thought to contribute to the organization of the intracellular space. However, also for proteins undergoing liquid de-mixing, the balance between function and dysfunction is far from clear. It is also unknown if, in vivo, liquid de-mixed states are precursors of insoluble amyloid-like states, and to which extent proteins are structured once in the liquid state.
How we do it
In order to understand how mutations affect these delicate equilibria and to elucidate when and why a sequence starts aggregating or becomes toxic for the cell, our lab integrates experimental and computational approaches in different model systems. Recently, we have developed massively parallel approaches based on Deep Mutational Scanning (DMS) to quantify the toxicity or the aggregation propensity of hundreds of thousands of protein sequences in vivo. We believe that by portraying the full landscape of the effects of mutations in a specific protein domain we can reach a more systematic and comprehensive understanding of the determinants of amyloid formation and toxicity.
We are also interested in developing similar high-throughput strategies to measure in vivo the effect of mutations on the physical state the proteins acquire upon mutation (diffuse, liquid de-mixed, insoluble) and to study the interactions between mutations to report on the conformations proteins adopt as they self-assemble. Overall, the exhaustive datasets we are generating will give mechanistic insights on the process of protein aggregation, while also reporting on specific conformations and mechanisms leading to cellular toxicity. We also aim at using the datasets we generate to develop novel predictors of protein aggregation.
We focus on all classical amyloids, such as the amyloid-beta peptide, the main component of the plaques found in Alzheimer’s disease patients, but also on functional amyloids and on a less characterized part of the human proteome which is able to undergo liquid de-mixing: prion-like domains. Just like all disordered protein regions, prion-like domains are particularly difficult to study in vitro. In this perspective, in vivo approaches such as the ones we develop, can provide a unique opportunity to investigate these sequences in a systematic way.
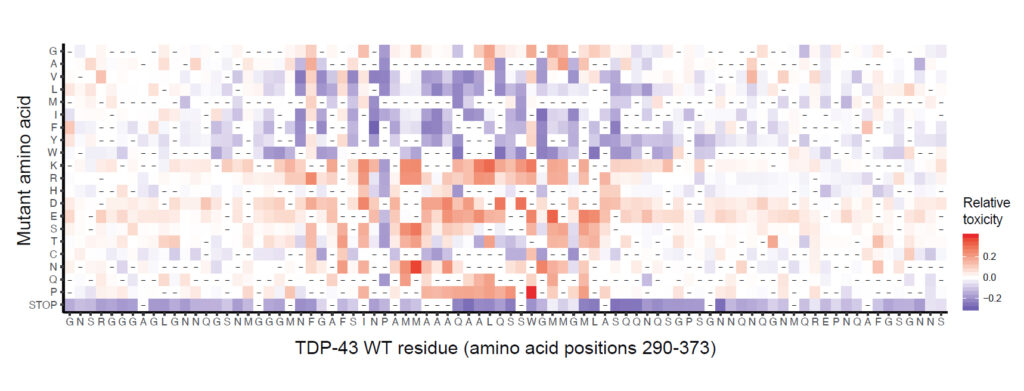
Map of the effect of mutations on toxicity of the TDP-43 Prion-like Domain.
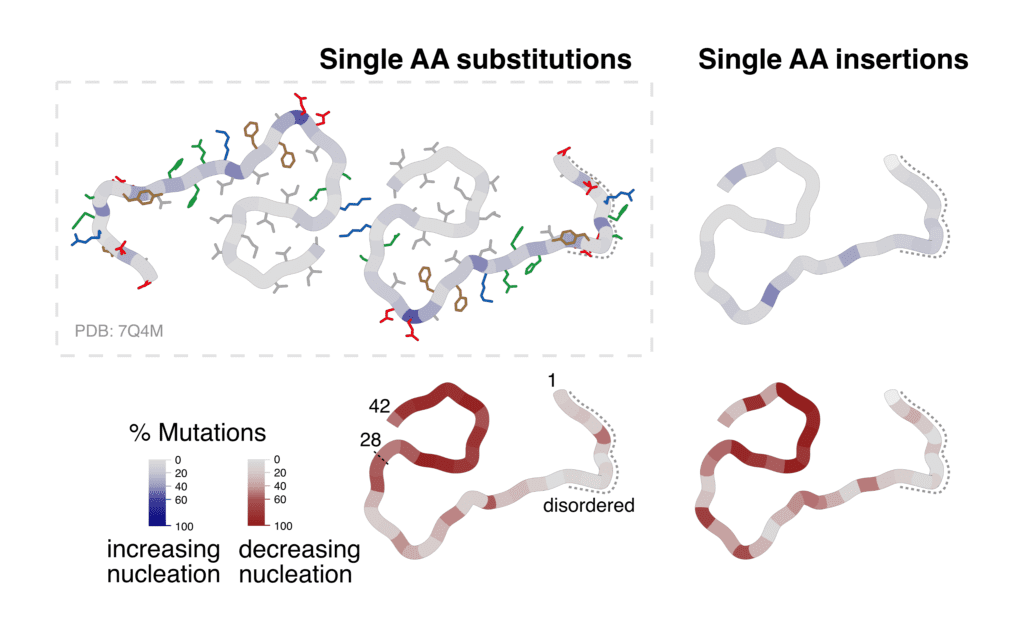
Percentage of substitutions and insertions increasing or decreasing amyloid formation of the Amyloid-Beta peptide, visualized on the cross-section of ex-vivo fibrils (7Q4M).
Staff
 ibecbarcelona.eu
ibecbarcelona.euProjects
| NATIONAL GRANTS | FINANCER | PI |
|---|---|---|
| AMYNDEL · Deciphering the consequences of different types of genetic variation in amyloid forming sequences by deep mutagenesis ( 2022-2025) | MICIU · Generación Conocimiento: Proyectos I+D | Benedetta Bolognesi |
| DeepAmyloids · Massively parallel mutagenesis to understand, predict and prevent amyloid nucleation in neurodegenerative diseases (2021-2024) | Obra Social La Caixa | Benedetta Bolognesi |
| FINISHED PROJECTS | FINANCER | PI |
|---|---|---|
| Poly-STOP · Developing modulators of protein aggregation in polyglutamine diseases by deep mutational scanning (2021-2022) | BIST · Barcelona Institute of Science and Technology | Benedetta Bolognesi |
| PRIOMUT · Escaneado exhaustivo de mutaciones en un dominio priónico para entender la toxicidad inducida por proteínas (2019-2021) | MICIU / Retos investigación: Proyectos I+D | Benedetta Bolognesi |
Publications
Equipment
- Thermo MaxQ 8000
Collaborations
- Priyanka Narayan
NIH-NIDDK - Xavier Salvatella
IRB, Barcelona - Fran Supek
IRB, Barcelona - Ben Lehner
CRG, Barcelona - Luke McAlary /Justin Yerbury
University of Wollongong, Australia
News
BIST Forum, un encuentro para poner en valor la investigación de frontera
El BIST Forum ha tratado de cómo la ciencia de excelencia potencia el desarrollo de la sociedad y el crecimiento económico. Han asistido el presidente de la Generalitat, el alcalde de Barcelona, los responsables de las máximas instituciones económicas y los rectores de las principales universidades. En el acto se han anunciado los nuevos proyectos BIST IGNITE para la investigación multidisciplinar, de los cuales tres cuentan con la participación del IBEC.
La investigadora Benedetta Bolognesi galardonada con una prestigiosa beca europea ERC Consolidator Grant
La investigadora del IBEC ha sido galardonada con una «ERC Consolidator Grant». Esta prestigiosa financiación europea respalda a científicos en la etapa de consolidación de sus equipos de investigación, permitiéndoles perseguir ideas científicas innovadoras. 2 millones de euros durante 5 años permitirán a Bolognesi y su equipo desarrollar un nuevo método para identificar las mutaciones que conducen a la formación de amiloides, agregados de proteínas que causan multitud de enfermedades, incluyendo el Alzheimer o el Parkinson.
Un nuevo atlas desvela qué mutaciones están detrás de la formación nociva de amiloide
Investigadores del Instituto de Bioingeniería de Cataluña (IBEC) han elaborado el atlas más completo hasta la fecha con las mutaciones genéticas que causan la formación de fibrillas de amiloide BETA, … Read more
Ciencia y Arte: el IBEC colabora en una obra de Antoni Muntadas en el Ars Electronica 2022
¿Cuándo se considera extinto un ser vivo? Esta es una de las cuestiones fundamentales de la obra de arte presentada en el Ars Electronica 2022 por Antoni Muntadas, artista catalán … Read more
Postdoctoral researcher at the Protein Phase Transitions in Health and Disease Research group
Ref: Postdoctoral / Deadline: 24th August
Lab Technician at the Protein phase transitions in health and disease Research Group
Ref: Lab Technician / Deadline: 29th July 2022
Investigadores del IBEC y CRG reciben financiación de la Fundación “la Caixa” para luchar contra la demencia
La Fundación “la Caixa” financiará un amplio e innovador proyecto de investigación codirigido por Benedetta Bolognesi, Junior Group Leader d en el IBEC, y por el profesor de investigación ICREA Ben Lehner del CRG, cuyo objetivo es conocer mejor las causas genéticas que conducen a enfermedades neurodegenerativas. Los investigadores combinarán técnicas de “mutagénesis profunda” y aprendizaje automático para producir un “mapa de la demencia” como método para predecir si una persona es más susceptible a padecer estas enfermedades.
Semillas científicas para salud: investigadores en IBEC ganan dos proyectos “Ignite Seed Grant” del BIST
Investigadores del IBEC reciben dos “Ignite Seed Grants” con los que combinarán sus capacidades con otros miembros del BIST para buscar respuestas científicas a retos en salud. Benedetta Bolognesi estudiará, con el IRB, la enfermedad de Huntington y otras patologías neurodegenerativas sin tratamiento. Por otro lado, Juan Manuel Fernández-Costa e investigadores en ICFO desarrollarán músculos-en-un-chip y sensores de biomagnetismo para acelerar el diseño de nuevos tratamientos de la distrofia muscular.
Un paso más hacia la detección precoz del alzhéimer
Benedetta Bolognesi, líder de grupo del IBEC, aparece en distintos medios por un reciente estudio publicado en la revista eLife. En el estudio muestran el primer mapa con miles de mutaciones en el gen que codifica el péptido beta amiloide para predecir qué personas son más propensas a desarrollar la enfermedad de Alzheimer.
El primer mapa completo de mutaciones en la placa amiloide abre nuevas vías para la detección temprana de la enfermedad de Alzheimer
Un estudio publicado en la revista eLife analiza todas las posibles mutaciones en el péptido beta amiloide para determinar cómo influyen en su agregación y formación placas, un sello patológico de la enfermedad de Alzheimer. También ayudará a los investigadores a comprender mejor los mecanismos biológicos que controlan la aparición de la enfermedad.
Jobs
Research Assistant at the Protein Phase Transitions in Health and Disease Research Group (RA_BB)
Ref: RA_BB // Deadline: 26/01/2024
Senior Laboratory Technician at the Protein Phase Transitions in Health and Disease Research Group (SLT_BB)
Ref: SLT_BB // Deadline: 11/01/2024
Research Assistant at the Protein Phase Transitions in Health and Disease Research Group
Ref: RA_BB // Deadline: 04/08/2023
Research Assistant at the Protein Phase Transitions in Health and Disease Research Group
Ref : RA_BB // Deadline 28/04/2023
Postdoctoral researcher at the Protein Phase Transitions in Health and Disease Research group
Ref: Postdoctoral / Deadline: 24th August
Lab Technician at the Protein phase transitions in health and disease Research Group
Ref: Lab Technician / Deadline: 29th July 2022
2 Postdoctoral researchers at the Protein Phase Transitions in Health and Disease Research Group
Application Deadline: 24/10/2021Ref: PD-BB The Protein phase Transitions in Health and Disease group at the Institute for Bioengineering of Catalonia (IBEC) is looking for two Postdoctoral Researchers to develop deep mutagenesis projects in the context of amyloid forming proteins. The contract will be within the framework of a larger project funded by La Caixa.