

About
Our lab aims at understanding how genetic changes between individuals can or cannot result in disease by quantifying the impact mutations have on protein aggregation and toxicity.
We are particularly interested in amino acid sequences that can adopt different conformations and undergo a process of self-assembly which results in distinct physical states.
Protein self-assembly
The aggregation of proteins into insoluble amyloid fibrils is a key process in the pathogenesis of a number of neurodegenerative conditions, such as Parkinson’s disease or Amyotrophic Lateral Sclerosis. However, examples of functional amyloid are also widespread in nature, especially across bacteria and fungi. Our work aims at systematically deciphering the sequence-dependencies of the process of aggregation in both functional and pathological contexts.
Proteins can also self-assemble into a more dynamic and reversible state through a process of condensation which is thought to contribute to the organization of the intracellular space. However, also for proteins that undergo liquid-demixing to form biomolecular condensates, the balance between function and dysfunction is far from clear. It is also unknown if and how condensates are precursors of insoluble amyloid-like states, and to which extent proteins are structured once in the liquid state.
Quantifying the impact of mutations at scale
In order to understand how mutations affect these delicate equilibria and to elucidate when and why a sequence starts aggregating or becomes toxic for the cell, our lab integrates experimental and computational approaches in different model systems. Recently, we have developed different Multiplexed Assays of Variant Effects (MAVEs) to quantify the toxicity and aggregation propensity of hundreds of thousands of protein sequences in vivo. By capturing the full landscape of the effects of mutations in a specific protein sequence we can guide clinicians to better diagnose and treat human disease, but we also reach a comprehensive mechanistic understanding of the process of amyloid formation and protein-induced toxicity. This translates in the possibility of rationally developing better targeted therapeutics, as well as in a set of fundamental principles that can guide protein design in bioengineering.
Developing novel strategies to report on protein conformation
In collaboration with the Lehner lab we also develop combinatorial mutagenesis approaches to study the interactions between mutations. We then use such genetic interactions to report on the conformation different proteins adopt as they start aggregating. Overall, we aim at generating exhaustive datasets that will give mechanistic insights on the process of protein aggregation, while also reporting on specific conformations and mechanisms leading to cellular toxicity. The massive amount of data we generate is used to train new models of protein aggregation. Our strategy is also amenable to tackle many intrinsically disordered proteins, which are particularly difficult to study in vitro. In this perspective, in vivo selection approaches such as the ones we develop can provide a unique opportunity to investigate these sequences in a systematic way.
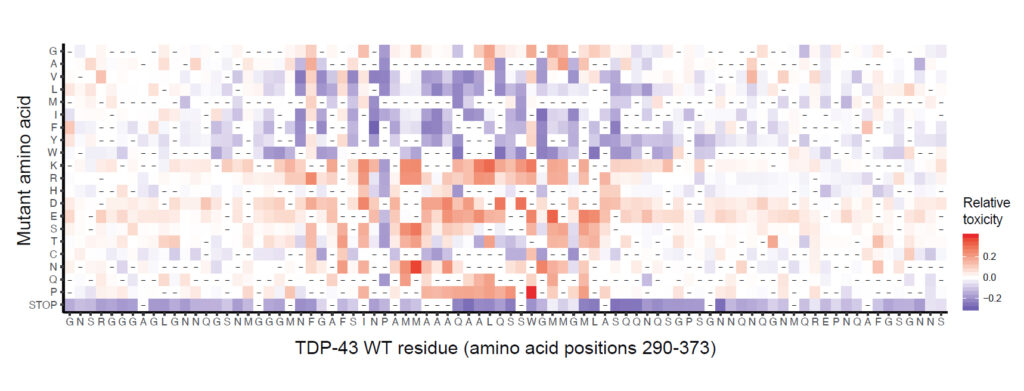
Map of the effect of mutations on toxicity of the TDP-43 Prion-like Domain.
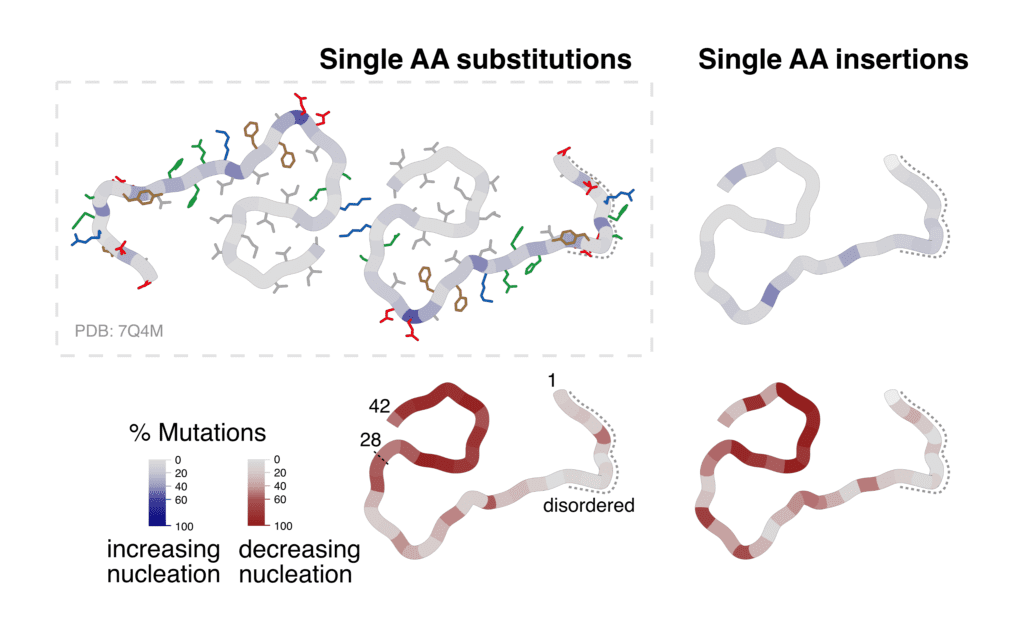
Percentage of substitutions and insertions increasing or decreasing amyloid formation of the Amyloid-Beta peptide, visualized on the cross-section of ex-vivo fibrils (7Q4M).
Staff
 ibecbarcelona.eu
ibecbarcelona.euProjects
| NATIONAL GRANTS | FINANCER | PI |
|---|---|---|
| AMYNDEL · Deciphering the consequences of different types of genetic variation in amyloid forming sequences by deep mutagenesis ( 2022-2025) | MICIU · Generación Conocimiento: Proyectos I+D | Benedetta Bolognesi |
| DeepAmyloids · Massively parallel mutagenesis to understand, predict and prevent amyloid nucleation in neurodegenerative diseases (2021-2024) | Obra Social La Caixa | Benedetta Bolognesi |
| FINISHED PROJECTS | FINANCER | PI |
|---|---|---|
| Poly-STOP · Developing modulators of protein aggregation in polyglutamine diseases by deep mutational scanning (2021-2022) | BIST · Barcelona Institute of Science and Technology | Benedetta Bolognesi |
| PRIOMUT · Escaneado exhaustivo de mutaciones en un dominio priónico para entender la toxicidad inducida por proteínas (2019-2021) | MICIU / Retos investigación: Proyectos I+D | Benedetta Bolognesi |
Publications
Check for more detailed information on the outputs of the Group at IBEC CRIS portal.
Publications list:
Equipment
- Thermo MaxQ 8000
Collaborations
- Priyanka Narayan
NIH-NIDDK - Xavier Salvatella
IRB, Barcelona - Fran Supek
IRB, Barcelona - Ben Lehner
CRG, Barcelona - Luke McAlary /Justin Yerbury
University of Wollongong, Australia
News

IBEC showcases cutting-edge biomedical innovation at the Health Revolution Congress 2026
The Institute for Bioengineering of Catalonia brought together researchers, investors and industry leaders with its event “IBEC Health Revolution Day: Engineering the Future of Medicine”, as part of the Health Revolution Congress. Through expert panels, technology pitches and scientific talks, the event showcased cutting-edge biomedical technologies and discussed how to translate scientific discoveries into real-world healthcare solutions.

An unprecedented mutation map reveals how amylin mutations influence type 2 diabetes
A study by the Institute for Bioengineering of Catalonia (IBEC) has created a mutational map of the hormone amylin, the aggregation of which can damage pancreatic cells and lead to type 2 diabetes. The team analysed 1,916 variants of the protein and compared the results of this mutational map with genetic and clinical data from 500,000 individuals. The study, published in Nature Communications, sheds new light on the role of genetics in diabetes and could lead to the development of safer and more effective drugs.

IBEC welcomes a new international cohort of doctoral students in collaboration with hospitals and centres of excellence
The Institute for Bioengineering of Catalonia (IBEC) is pleased to welcome new international students to its PhD programme. This initiative offers excellent training and research opportunities in bioengineering, as well as strong collaboration with leading hospitals and institutions.

Three IBEC projects receive ERC Proof of Concept funding to drive innovation in health, neuroscience, and biomedical technology
Dr Benedetta Bolognesi, Dr Irene Marco-Rius and Dr Nicolò Accanto, who are all principal investigators at IBEC, have each been awarded a prestigious ERC Proof of Concept grant. These grants are awarded by the European Research Council to support the exploration of the commercial and societal potential of research projects conducted within European institutions. The three winning projects range from new platforms for discovering antiamyloid drugs, to advanced metabolic analysis technologies, to optical tools for studying the brains of animals moving naturally.

Bioengineering for precision medicine at the 18th IBEC Symposium
The 18th annual IBEC Symposium focused on ‘Bioengineering for Precision Medicine’, which is one of IBEC’s key areas of application. The event was attended by nearly 300 people, including local and international researchers. The multidisciplinary environment provided experts from other centres and the IBEC community with the opportunity to present their projects and exchange knowledge.

IBEC and EMBL Barcelona co-organise a matchmaking workshop to explore synergies
Today, the Institute for Bioengineering of Catalonia (IBEC) and the European Molecular Biology Laboratory (EMBL) held a matchmaking workshop. The event brought together leading researchers from both centres to encourage the formation of new connections and promote scientific dialogue.

IBEC and Antoni Muntadas featured in the ‘Invisible Animals’ exhibition at the Natural Science Museum of Barcelona
The Institute for Bioengineering of Catalonia (IBEC) attended the opening of the exhibition, which highlights extinct fauna that remains present in the collective imagination. The exhibition includes a section dedicated to the Tasmanian tiger featuring a piece by Antoni Muntadas, on which IBEC collaborated with Benedetta Bolognesi, a principal investigator at the institute.

Seven additional IBEC labs achieve top-level in My Green Lab certification
Seven more research groups at the Institute for Bioengineering of Catalonia (IBEC) have been certified by My Green Lab, reaching the highest rating, the Green Level, for sustainable laboratory practices. With these additions, IBEC core facilities and 70% of the Institute’s laboratories are now certified.

IBEC and the Hospital del Mar formalise a new collaboration
The first collaboration day between the Institute for Bioengineering of Catalonia (IBEC) and the Hospital del Mar Research Institute was held today at the Barcelona Biomedical Research Park (PRBB). The meeting provided an opportunity to share research projects, identify areas for collaboration and formalise a strategic alliance through the signing of a collaboration agreement between IBEC, the Hospital del Mar and the Hospital del Mar Research Institute.

Scientists map the first step in Alzheimer’s protein aggregation and discover clues for future therapies
This is an analysis on an unprecedented scale. They studied over 140,000 versions of the Aβ42 peptide, which forms harmful plaques in the brain. It is the first map to reveal how mutations affect a protein in its transition state — a fleeting phase that is difficult to study. This finding opens up new avenues for preventing Alzheimer’s disease and suggests a method that can be applied to studying other proteins involved in different pathologies. The study, published in Science Advances, is a collaboration between the Wellcome Sanger Institute in the United Kingdom, the Institute for Bioengineering of Catalonia, and the Centre for Genomic Regulation in Barcelona.
Jobs
Post-doctoral Fellow at the Phase Transitions in Health and Disease Research Group
Ref: PR-BB // Deadline: 23/12/2025
Laboratory Techncian at the Phase Transitions in Health and Disease Research Group
Ref: LT-BB // Deadline: 9/12/2025
Senior Laboratory Technician at the Protein Phase Transitions in Health and Disease Research Group
Ref: SLT-BB // Deadline: 8/12/2025
Laboratory Technician in the Phase Transitions in health and disease Research Group
Ref: LT-BB// Deadline: 28/09/2025
Researcher in Training at the Phase Transitions in Health and Disease Research Group
Ref: RT-BB // Deadline: 30/07/2025
Research Assistant at Protein Phase Transitions in Health and Disease Research Group
Ref: RA-BB //Deadline : 16/01/2025
Research Assistant at the Protein Phase Transitions in Health and Disease Research Group
RA-BB // Deadline: 16/09/2024
Predoctoral researcher at the Phase Transitions in Health and Disease Research Group
Ref: PHDR_BB // Deadline: 12/08/2024
Post-doctoral researcher in the Protein Phase Transitions in Health and Disease Research Group
Ref: PR_BB // Deadline 30/10/2024
Predoctoral researcher at the Phase Transitions in Health and Disease Research Group
BB_PR // Deadline: 25/07/2024